Authors: Dr. Daniel J. Arismendi‐Arrieta, Dr. Álvaro Valdés, Dr. Rita Prosmiti.
Contribution: Article
Journal: Chem. Eur. J.
Publication date: 24, 9353-9363 (2018).
Abstract:
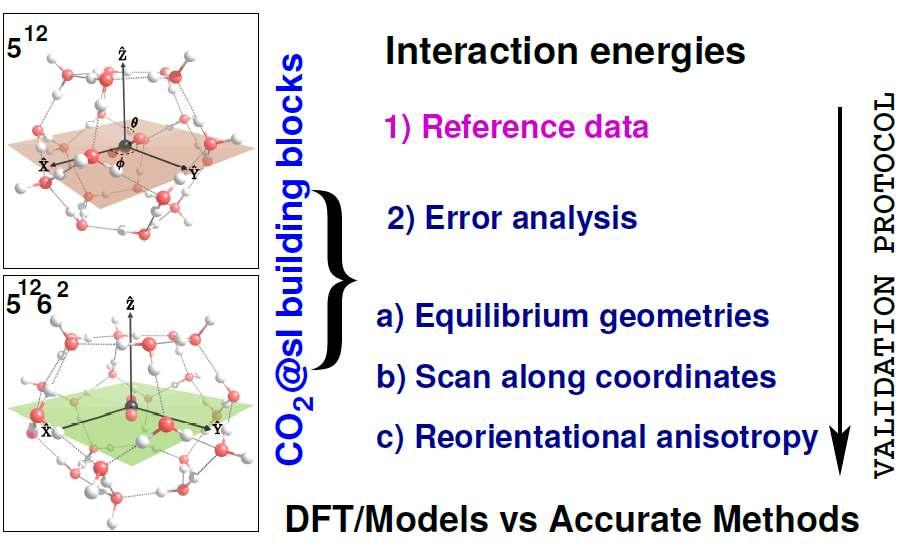
Clathrate hydrates of CO2 have been proposed as potential molecular materials in tackling important environmental problems related to greenhouse gases capture and storage. Despite the increasing interest in such hydrates and their technological applications, a molecular‐level understanding of their formation and properties is still far from complete. Modeling interactions is a challenging and computationally demanding task, essential to reliably determine molecular properties. First‐principles calculations for the CO2 guest in all sI, sII, and sH clathrate cages were performed, and the nature of the guest–host interactions, dominated by both hydrogen‐bond and van der Waals forces, was systematically investigated. Different families of density functionals, as well as pairwise CO2@H2O model potentials versus wavefunction‐based quantum approaches were studied for CO2 clathrate‐like systems. Benchmark energies for new distance‐dependent datasets, consisting of potential energy curves sampling representative configurations of the systems at the repulsive, near‐equilibrium, and asymptotic/long‐range regions of the full‐dimensional surface, were generated, and a general protocol was proposed to assess the accuracy of such conventional and modern approaches at minimum and non‐minimum orientations. Our results show that dispersion interactions are important in the guest–host stabilization energies of such clathrate cages, and the encapsulation of the CO2 into guest‐free clathrate cages is always energetically favorable. In addition, the orientation of CO2 inside each cage was explored, and the ability of current promising approaches to accurately describe non‐covalent CO2@H2O guest–host interactions in sI, sII, and sH clathrates was discussed, providing information for their applicability to future multiscale computer simulations.