Authors: Castro-Gomez, L. Beatriz; Campos-Martínez, José; Hernández, Marta I.; Hernandez-Lamoneda, Ramon
Journal: CHEMPHYSCHEM
Publication date: 2023/10/05
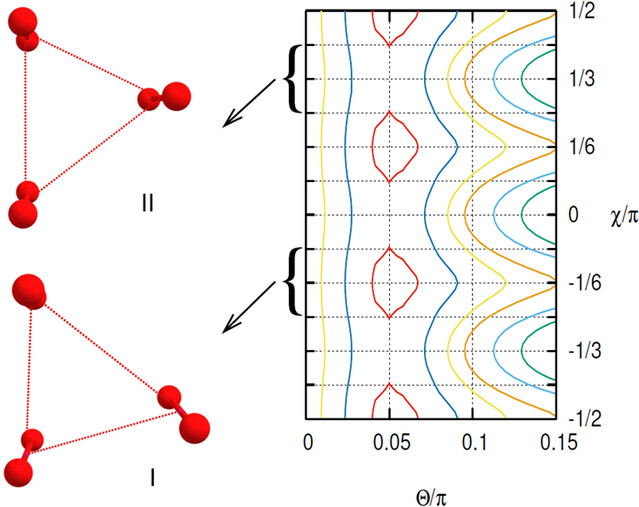
Abstract: We present a detailed theoretical study of the molecular oxygen trimer where the potential energy surfaces of the seven multiplet states have been calculated by means of a pair approximation with very accurate dimer ab initio potentials. In order to obtain all the states a matrix representation of the potential using the uncoupled spin representation has been applied. The S = 0 and S = 1 states are nearly degenerate and low-lying isomers appear for most multiplicities. A crucial point in deciding the relative stabilities is the zero-point energy which represents a sizable fraction of the electronic well-depth. Therefore, we have performed accurate diffusion Monte Carlo studies of the lowest state in each multiplicity. Analysis of the wavefunction allows a deeper interpretation of the cluster structures, finding that they are significantly floppy in most cases.